本文發表於《大眾科學》的前部落格網路,僅反映作者的觀點,不一定反映《大眾科學》的觀點
微生物無處不在,更不用說在我們體內了,但是發現和描述它們並非易事。但事情正在變得容易。正如我去年寫到的,DNA測序技術的進步意味著我們不再侷限於分離單個微生物,在實驗室中培養它們並逐個研究。相反,我們可以從特定環境中分離DNA,然後對其中存在的所有物質進行測序。但是,新期刊《自然·微生物學》上發表的一篇論文表明,分析新微生物種群的最常用方法系統地遺漏了其中很大一部分。
除非您是微生物學家,否則您可能需要了解標題中的兩個術語——“宏基因組學”和“基於擴增子的檢測”的入門知識。我在去年的系列文章中對它們進行了更完整的介紹,但我將簡要回顧一下。對於這種測序方法,一個有用的比喻是走進圖書館並嘗試編目其內容。擴增子測序就像掃描書脊並僅記錄其標題,而宏基因組測序就像瀏覽並記錄所有書籍內的文字。當然,您可以想象哪種方法更容易。
關於支援科學新聞報道
如果您喜歡這篇文章,請考慮透過以下方式支援我們屢獲殊榮的新聞報道 訂閱。透過購買訂閱,您正在幫助確保有關塑造我們當今世界的發現和想法的具有影響力的故事的未來。
這個比喻並不完美,因為基因組沒有容易訪問的“標題”。相反,科學家們知道標題通常採用的形式,並且可以使用稱為聚合酶鏈反應或PCR的技術來擴增這些序列。這篇論文表明,科學家們一直在使用的所謂“通用”引物並不像我們想象的那麼通用。這就像我們只一直在尋找以字母開頭的標題,並且在許多情況下完全錯過了奧威爾的《1984》。
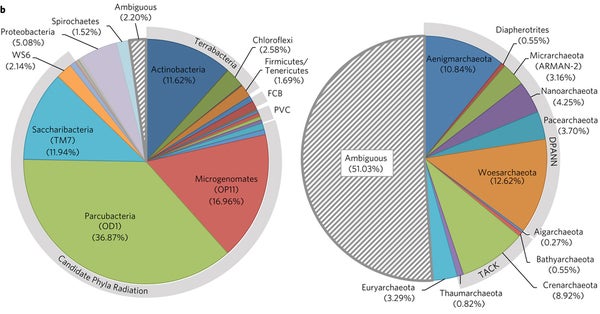
Emiley A. Eloe-Fadrosh 及其同事分析了超過 6000 個宏基因組資料集,並檢查了常用微生物引物可以檢測到多少 SSU 基因(用作基因組標題的基因)。他們發現,10% 或更多會被完全遺漏。毫不奇怪,大多數會被遺漏的細菌是新型或至少研究非常不足的物種的成員。

圖 1a - 給定引物會遺漏的宏基因組樣本中微生物的百分比。
不幸的是,解決這個問題的方法並不明顯。作者指出了單細胞和深度宏基因組測序,但是這些技術仍然相對昂貴,尤其是在分析返回的大量資料所需的專業知識和計算基礎設施方面。在功能性地描述這些大量的新細菌方面也存在巨大差距——僅僅因為我們知道它們在那裡,並不意味著我們知道它們是做什麼的。儘管如此,知道它們在那裡是必要的第一步。