
Susan 還是個孩子的時候,就首次懷疑母親可能出了問題。母親在做晚餐或洗碗時,杯子或盤子經常會意外掉到地上。“她會說自己‘笨手笨腳’,但她其實並不笨手笨腳,”Susan 說。“她的手部動作優美而富有魅力,我現在才意識到那是亨廷頓舞蹈症(HD)的早期症狀。”
亨廷頓舞蹈症(HD)是一種遺傳性疾病,會導致大腦廣泛退化,並擾亂思維、行為、情緒和運動。這種疾病通常在中年開始,最初表現為情緒波動和難以集中注意力等細微變化。隨著病情發展,患者會發展為痴呆症,並喪失說話或行動能力。
Susan 要求隱瞞她的姓氏以保護她的隱私,她清楚地記得得知母親患病的那一天。那是 1982 年的春天,她的母親因極度疲憊、頻繁跌倒和不規則運動而住院。當時還沒有針對該疾病的基因檢測,因此她接受了一系列評估。她的神經科醫生將全家人聚集到一個房間裡宣佈了這個訊息。“他告訴我們,我們的母親患有亨廷頓舞蹈症,”Susan 回憶道。“而且沒有治療方法,如果你們不生育,它可能會在一代人內消失。”
支援科學新聞報道
如果您喜歡這篇文章,請考慮透過以下方式支援我們屢獲殊榮的新聞報道 訂閱。透過購買訂閱,您將幫助確保未來能夠繼續報道關於塑造我們當今世界的發現和思想的具有影響力的故事。
這些直率的話語對 Susan 和她的兄弟姐妹的生活產生了深遠的影響:她的哥哥決定永遠不結婚,她的姐姐選擇絕育。然而,對於 Susan 來說,這些選擇都遙不可及:當她收到訊息時,她已經懷孕了。
Susan 說,她和她的丈夫“無法決定什麼是正確的做法”。她特別想到的是,“如果我們生下這個孩子,那麼這個孩子長大後也會面臨同樣的決定”,她說。“這似乎太殘酷了。” 最終,這對夫婦做出了令人心碎的選擇,終止了妊娠。
參與亨廷頓舞蹈症的基因名為 HTT,它編碼一種名為亨廷頓蛋白的蛋白質。該基因的缺陷版本會多次重複其序列的一小段——核苷酸組合 CAG。與某些遺傳疾病不同,在某些遺傳疾病中,一個人只有擁有兩個缺陷基因副本才會患病,而僅一個 HTT 突變副本就足以導致亨廷頓舞蹈症,並且突變攜帶者有 50% 的機率將其遺傳給子女。在 Susan 的母親去世多年後,這三兄妹發現他們都遺傳了這種疾病。

圖片來源: Nik Spencer Nature
儘管亨廷頓舞蹈症的遺傳病因自 1993 年以來已經明確,但目前尚無治療方法可以阻止或減緩其進展。大多數其他神經退行性疾病也缺乏有效的療法,儘管它們的遺傳根源不如亨廷頓舞蹈症那樣明確,但與運動神經元疾病(肌萎縮側索硬化症,或 ALS)、阿爾茨海默病和帕金森病等疾病相關的許多基因已經為人所知幾十年了。現在,治療這類疾病的潮流可能正在轉變。許多研究人員對稱為反義寡核苷酸(ASO)的藥物寄予厚望。這些是由 DNA 或 RNA 字母組成的一小段鏈,旨在附著在缺陷基因產生的特定 RNA 序列上,並重新平衡它們產生的蛋白質水平——增加缺失的蛋白質或抑制缺陷蛋白質(參見“調節問題蛋白質”)。
美國食品藥品監督管理局(FDA)於 2016 年批准了首個用於神經系統疾病的 ASO,此後該領域的活動呈爆炸式增長。該領域已經從過去二十年中進行的少數臨床試驗發展到目前正在進行的約十幾個針對各種神經退行性疾病的臨床試驗——其中一些已經進入最後階段。
其他 ASO 研究人員正在超越由單一突變定義的疾病,開始研究具有更復雜遺傳基礎的疾病。最近的進展使該領域的許多人對這項技術的未來感到樂觀。加州大學聖地亞哥分校(UCSD)的神經科學家 Don Cleveland 是最早研究使用 ASO 治療神經系統疾病的科學家之一,他認為這僅僅是開始。“未來還有更多、更多,”他說。
但該領域的進展並非完全順利。上個月底,一項大型III 期臨床試驗突然中止,因為該藥物對患者的益處不足以抵消風險。一些研究人員長期以來一直敦促對 ASO 保持謹慎,因為它們在許多疾病中的療效尚不清楚,而且它們的給藥方式——通常是透過脊髓注射——具有侵入性。
雖然這項試驗的結果令人失望,但“我不認為這是絕望的理由,”位於馬里蘭州貝塞斯達的美國國家神經疾病與中風研究所負責監督基因療法的科學專案經理 Chris Boshoff 說。“我們仍然有理由對這種療法能夠取得的成就保持積極和熱情。”
罕見疾病的突破
Elliot 和 Janell Lewis 的第一個孩子 Blakely 於 2011 年出生,患有罕見的遺傳性神經退行性疾病,即脊髓性肌萎縮症(SMA)。SMA 患者攜帶 SMN1 基因的突變形式,該基因負責產生一種名為運動神經元存活蛋白(SMN)的蛋白質。由此導致的 SMN 缺乏會阻止大腦與身體進行有效溝通,導致肌肉無力和萎縮,並且隨著時間的推移會惡化。SMA 有四種類型;最常見的形式 SMA1 也是最嚴重的。SMA1 患者通常在出生後不久出現症狀,許多人活不過兩歲。
Blakely 在三個月大的時候被診斷出患有 SMA。“那幾乎擊垮了我們,”Elliot 說。當時,沒有治療方法,Blakely 在 21 個月大時去世了。
2017 年春天,這對夫婦又生了一個女兒 Evie。Evie 也患有 SMA,但她更幸運——在她出生前幾個月,FDA 批准了一種 ASO,名為 nusinersen,這是有史以來第一個改變 SMA 病情的治療方法。Evie 在出生 12 天時接受了她的第一劑藥物。
%20SMA1035.jpg?w=2000)
四歲的 Evie Lewis 在猶他州奧格登的家中玩耍。 圖片來源:Kim Raff Nature
科學家們早在 1978 年就認識到 ASO 靶向 RNA 的能力,但花了數十年才證明其臨床潛力。早期,毒性和效力不足等問題阻礙了進展,許多製藥公司失去了興趣。但是,位於加利福尼亞州卡爾斯巴德的一家公司 Ionis Pharmaceuticals(原名 Isis Pharmaceuticals)的研究人員對藥物的化學骨架進行了關鍵修改,提高了效力和穩定性,使 ASO 能夠到達靶點而不會被降解。
導致 nusinersen 誕生的工作始於 2000 年左右,地點在紐約的冷泉港實驗室,生物化學家和分子遺傳學家 Adrian Krainer 在那裡研究 SMN2(另一個編碼 SMN 的基因)通常比其對應物 SMN1 產生更少活性蛋白質的機制。他們推斷,如果他們能讓 SMN2 產生更多蛋白質,就可以彌補攜帶該基因突變的人體內 SMN1 的不足。他們從其他人的研究中瞭解到,幾乎在所有人中,SMN2 問題的根源在於剪接過程中的錯誤——RNA 鏈被剪下並加工成製造蛋白質的指令的過程。這會導致 SMN2 程式碼的一部分被跳過。
Krainer 的團隊專注於與 RNA 鏈結合並導致片段丟失的蛋白質,希望阻止它們干擾完整 SMN 蛋白質的生成過程。2004 年,Krainer 開始與藥理學家 Frank Bennett 合作,後者是 Ionis Pharmaceuticals 的創始成員之一。他們共同確定了一種 ASO,它可以與 RNA 鏈結合,並將該片段隱藏起來,使其免受沉默蛋白的影響,從而實現功能性 SMN 的生產。
該化合物 nusinersen 於 2011 年進入臨床試驗。結果非常喜人,以至於針對 SMA 嬰兒的 III 期試驗提前終止:接受該藥物的患者比接受安慰劑的患者更有可能達到他們的運動里程碑並存活下來。
迄今為止,全球已有超過 10,000 人接受了 nusinersen(Spinraza)治療,Ionis 於 2016 年將其授權給位於馬薩諸塞州劍橋市的製藥商 Biogen。該藥物徹底改變了疾病的程序:出生後不久接受治療的 SMA 嬰兒不再在生命的最初幾年內死亡。如今,“(與家人的)對話不再僅僅以‘我們將盡一切努力,但您的寶寶還是會死’結束,”猶他大學兒科神經科醫生 Russell Butterfield 在鹽湖城說道(Butterfield 收到了 Biogen 的諮詢費)。“相反,對話轉變為,‘我們有這種新藥,它絕對令人驚歎。我們需要儘快給孩子用上’。”
Evie Lewis 現在四歲了,她每隔幾個月透過腰椎穿刺接受一次 Spinraza 注射,最近她接受了第 15 次注射。儘管她仍然面臨一些問題,例如必須透過飼管進食,但她能夠走路、跑步和攀爬——這些都是 Blakely 從未能夠做到的事情,Elliot 說。
擁擠的研發管線
繼 nusinersen 成功之後,研究人員開始著手解決其他與明確定義的基因突變相關的疾病,例如亨廷頓舞蹈症。這促成了藥物 tominersen 的誕生,該藥物由 Ionis 開發,並授權給瑞士巴塞爾的製藥公司 Roche 進行臨床試驗。據認為,它的作用機制是靶向正常和缺陷 HTT 基因產生的 RNA 鏈上的 CAG 重複序列,並標記它們以便被一種名為 RNase H1 的酶破壞。2019 年公佈的 I/II 期臨床試驗結果顯示,tominersen 降低了腦脊液中突變型亨廷頓蛋白的濃度,且未引起任何嚴重的副作用。
早期亨廷頓舞蹈症試驗的成功引起了神經退行性疾病研究人員的關注,因為蛋白質纏結是許多此類疾病的關鍵特徵。“對此感到非常興奮,因為它真正打開了為其他毒性突變蛋白積累起作用的神經退行性疾病進行反義試驗的大門,”倫敦大學學院的神經科醫生 Sarah Tabrizi 說,她是 tominersen 的 I/II 期試驗的負責人。
但 3 月底的一項意外公告給亨廷頓舞蹈症群體帶來了沉重打擊。一項涉及來自 18 個國家的 791 名參與者的 tominersen III 期試驗,根據獨立專家委員會的建議提前終止,該委員會對資料進行了計劃中的審查。羅氏公司的一份宣告稱,沒有出現新的安全問題,但該藥物的潛在益處不足以抵消風險。Tabrizi 說,在更多細節公佈之前,無法確定哪裡出了問題。
以與 tominersen 類似方式起作用的藥物仍在用於其他具有相似病因的疾病。例如,某些 ALS 病例是由過多的突變蛋白引起的,並且針對這些疾病形式的一些 ASO 正在進行臨床試驗。進展最快的是 tofersen,一種由 Ionis 開發的 ASO,用於治療一種遺傳性 ALS。Tofersen 目前正在Biogen 贊助的 III 期試驗中進行測試。
弗吉尼亞聯邦大學裡士滿分校的神經科醫生 Claudia Testa 說,與像 nusinersen 那樣促進缺失蛋白質相比,像 tominersen 和 tofersen 那樣降低突變蛋白質水平會帶來獨特的挑戰。一些降低蛋白質的策略實際上會降低蛋白質的良好和不良版本的水平。科學家們尚不清楚對相關疾病的長期影響,也不清楚這是否是 tominersen III 期試驗中的問題。用於 SMA 的藥物正在做一些根本不同的事情,“因此它不能預測其他疾病的療效——這是一個痛苦的事實”,Testa 說。
為了避免這個問題,一些 ASO 的目標直接針對突變蛋白。位於馬薩諸塞州劍橋市的一家生物技術公司 Wave Life Sciences 正在測試一種策略,該策略針對有時與 HTT 突變副本上的 CAG 重複序列同時發生的小突變。目的是使健康亨廷頓蛋白的水平保持相對完整。但該藥物僅對攜帶這些突變的亨廷頓舞蹈症患者的子集有效。此外,Testa 說,只有透過一種詳盡的測序方法才能識別出這種差異,而這種方法在臨床上並未常規進行。(Testa 收到了 Wave Life Sciences 的諮詢費。)
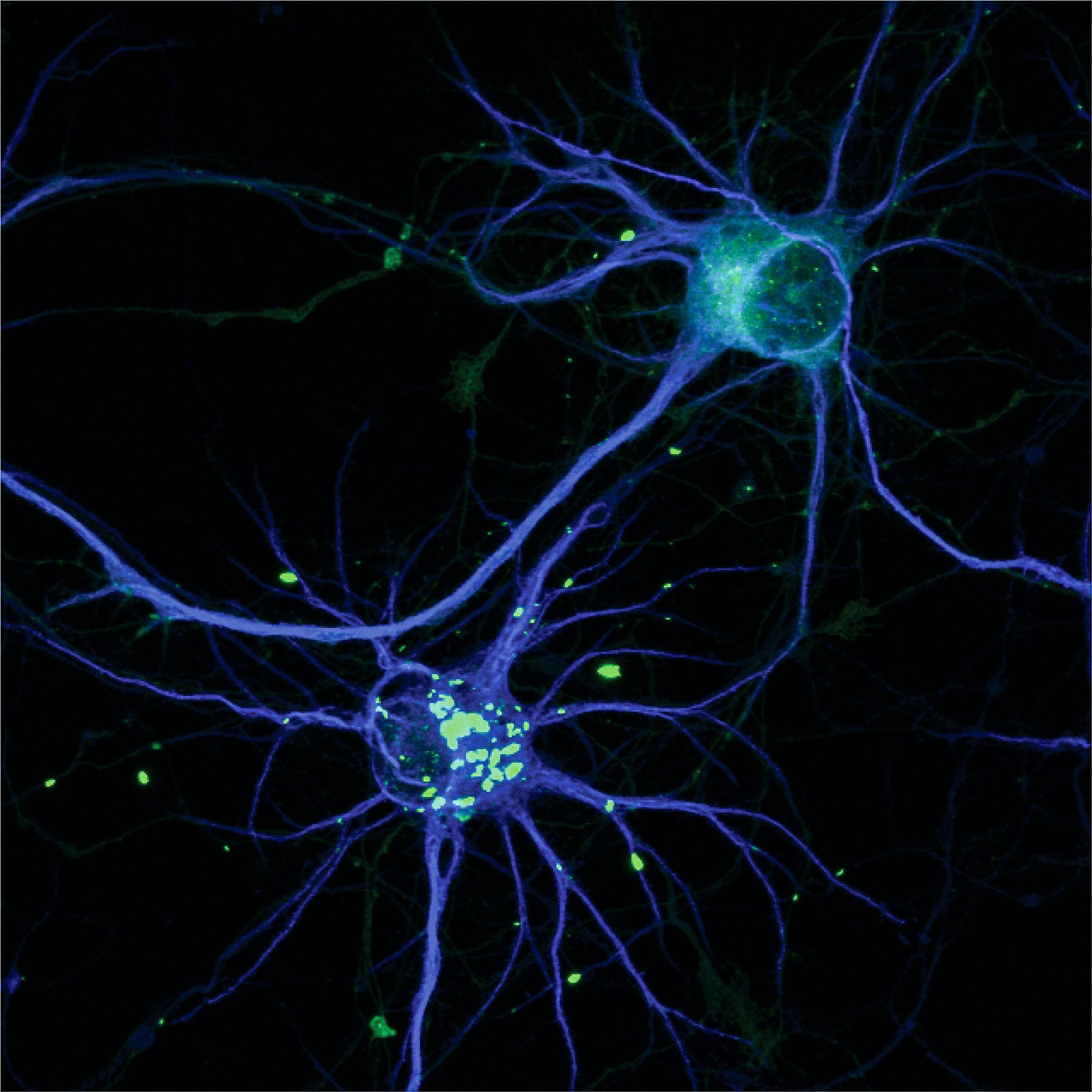
導致亨廷頓舞蹈症的 HTT 蛋白的毒性版本,形成團塊(亮綠色)。 圖片來源:K. W. Drombosky 等人。
最近,研究人員已開始測試基於 ASO 的療法,用於更常見的神經退行性疾病,例如帕金森病和阿爾茨海默病。絕大多數病例與特定的基因突變無關,而且這些疾病比遺傳性疾病更為普遍。用於阿爾茨海默病的 ASO 旨在降低 tau 蛋白的水平,tau 蛋白會在大腦中積聚成有毒的纏結。對於帕金森病,目標是降低 α-突觸核蛋白,該蛋白會聚集形成稱為路易體的病理團塊。
但對於此類神經退行性疾病,一個網路中的多個基因可能都參與其中,英國牛津大學的神經科醫生 Kevin Talbot 說,他將參與即將進行的 ALS 的 ASO 試驗。他說,尚不清楚網路中一個基因的變化將如何影響其餘基因。(Talbot 曾擔任 Roche 和 Biogen 的科學顧問委員會成員。)
Talbot 認為,另一個問題是,這些藥物目前需要透過重複的腰椎穿刺才能到達它們在中樞神經系統中的靶點。Talbot 說,在 ASO 可以應用於更廣泛的疾病之前,重要的是找到一種方法讓這些藥物穿過血腦屏障,從而以侵入性更小的方式給藥。“在我們變得過於樂觀之前,還有很多事情要做。”
身份轉變
小鼠研究表明,未來的 ASO 可能在大腦中發揮更強大的作用:替換丟失的神經元。
去年,加州大學聖地亞哥分校的細胞生物學家付向東和他的同事們證明,可以使用 ASO 將稱為星形膠質細胞的非神經元腦細胞轉化為神經元。該團隊將 ASO 注射到帕金森病小鼠大腦中神經元丟失的區域。一旦到達那裡,該藥物就會啟用一個基因網路,促使星形膠質細胞變成神經元。在帕金森病小鼠模型中,付向東的團隊發現,接受治療的動物在某些行為方面有所改善。
參與了付向東試驗的 Cleveland 一直與 Ionis 提供的 ASO 合作,以在大腦的其他部位測試這個想法。“這真的是我將投入我職業生涯剩餘時間的地方,”他說。“我確信我們才剛剛開始思考這種可能性。”
這些星形膠質細胞轉化 ASO 仍處於早期階段。付向東警告說,在將這項技術應用於臨床之前,需要在非人類靈長類動物身上進行測試,因為它們的 Brains 比小鼠更接近我們自己的大腦。
目前,研究人員正在熱切等待 tofersen ALS III 期試驗的結果,以及關於 tominersen 亨廷頓舞蹈症試驗為何中止的更多資訊。
Susan 是一位 60 多歲的退休護士,自 I 期以來就參與了 tominersen 試驗。她說,她對這個訊息感到失望,但她感謝作為參與者所受到的照顧。“我很榮幸從第一天起就參與了這項試驗。現在只是耐心和審查的問題。沒有其他選擇,不是嗎?”
本文經許可轉載,並於 2021 年 4 月 6 日首次釋出。