
以下文章經 The Conversation許可轉載,The Conversation是一家報道最新研究的線上出版物。
在經過14年多的研發後,兩名嬰兒接受了針對泰-薩克斯病的首個基因療法。
支援科學新聞報道
如果您喜歡這篇文章,請考慮透過以下方式支援我們屢獲殊榮的新聞報道: 訂閱。透過購買訂閱,您將幫助確保有關塑造我們當今世界的發現和思想的具有影響力的故事的未來。
泰-薩克斯病是一種嚴重的神經系統疾病,由一種名為HexA的酶缺乏引起。這種酶分解一種通常以非常少量、無害的量存在於大腦中的類脂肪物質。然而,如果沒有HexA,這種類脂肪物質會積累到毒性水平,從而損害和殺死神經元。
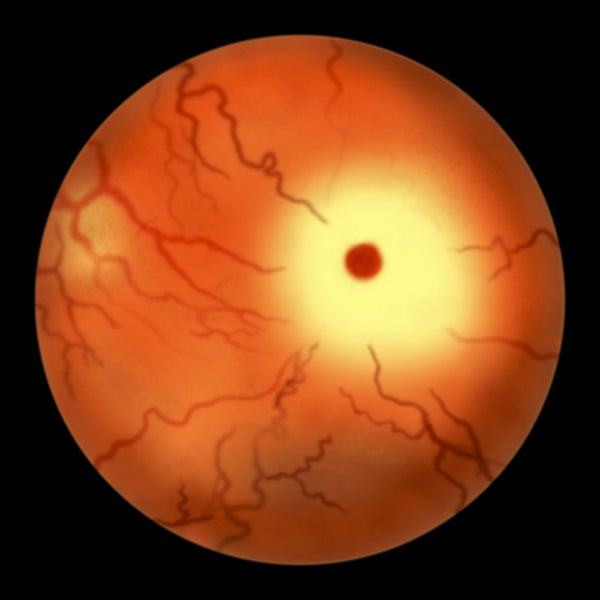
英國眼科醫生沃倫·泰於1883年首次描述了該疾病的症狀之一,他在受影響的嬰兒的眼睛後部看到了櫻桃紅斑。1887年,美國神經學家伯納德·薩克斯在一篇開創性的論文中描述了泰-薩克斯病深刻的神經系統症狀
“……直到兩到三個月大的時候,才注意到任何異常,當時父母觀察到孩子比同齡的孩子更加無精打采。……孩子通常會仰臥,並且永遠無法改變姿勢……它從未嘗試任何自主運動……孩子變得越來越虛弱,它停止正常進食,它的支氣管問題加劇,最後,肺炎發作,它於1886年8月去世。”
這種對泰-薩克斯病的令人沮喪的描述仍然適用,患有該疾病的人通常在5歲前死亡。有些人會在晚年患上泰-薩克斯病,症狀在青少年時期開始出現,並在幾十年內逐漸惡化。
不幸的是,泰-薩克斯病仍然沒有治療方法。積極的醫療治療可以延長生存期,但不能改善神經功能。治療泰-薩克斯病的唯一有效方法是恢復大腦中的HexA酶。然而,這很困難,因為血腦屏障阻止了大多數分子進入大腦。
我是來自麻省大學陳醫學院和奧本大學的研究人員團隊的成員,我們開發了一種基因療法,可能有助於繞過這一屏障。我們的治療方法使用兩種無害的病毒載體將DNA指令傳遞給腦細胞,教導它們如何產生缺失的酶。類似的技術已被用於治療多種相關疾病和其他疾病。對於泰-薩克斯病,這些DNA指令進入這些細胞的細胞核並停留在那裡,從而可以長期產生HexA。根據我們之前在不同動物物種身上成功測試我們的基因療法的研究,我們認為將治療方法輸送到大腦的中心部分可以讓酶沿著其連線傳播到其他區域,並分佈到整個大腦。
第一個接受我們基因療法治療的兒童年齡為2歲半,患有晚期疾病症狀。治療三個月後,他們的肌肉控制能力更好,並且可以集中注意力。現在5歲時,該兒童健康狀況穩定且無癲癇發作,這對於這個年齡段的患者來說通常是不可能的。第二個在7個月大時接受治療的兒童在三個月的隨訪中大腦發育有所改善,並且在2歲多一點時仍然沒有癲癇發作。
需要進行更多測試以確認我們的治療方法是否可以完全阻止疾病進展。鑑於這是我們的治療方法首次應用於人體,我們使用了低於我們在動物研究中看到的最高治療效果的保守劑量。我和我的同事目前正在進行一項後續臨床試驗,以測試在更多患者中增加劑量的安全性和有效性。
研究罕見疾病可以促進整個醫學的進步。
這些療法不斷上漲的製造成本使得為許多全球患者人數非常少且盈利能力低的超罕見疾病開發和測試基因療法變得極其困難,甚至不可能。
我們能夠為正在進行的臨床試驗中的兒童提供這些治療,這完全歸功於一個慷慨的家庭的資助,他們自己的孩子也是參與者。這種基層方法是超罕見疾病研究中的一個常見主題——開發和測試通常由父母、基金會和聯邦撥款支援。
我們在麻省大學陳醫學院的轉化分子治療研究所計劃致力於與家庭和基金會合作,為越來越多的超罕見疾病開發更多的病毒載體基因療法。我們相信,全世界大約7,000種罕見疾病中的每位患者都應該有機會過上正常的生活。
本文最初發表於 The Conversation。閱讀 原文。